Lynch syndrome genetics and clinical implications
The Gastroenterology journal (January 2023) features a review article by Professors Päivi Peltomäki, Minna Nyström, Jukka-Pekka Mecklin, and Toni T. Seppälä titled “Lynch Syndrome Genetics and Clinical Implications”. The article presents the recent advances in understanding the molecular basis for Lynch syndrome-associated tumor development and describes their implications for clinical management. The aim is to review how the progress in the Lynch syndrome (LS) field has contributed to a more complete picture of LS, resulting in strategies for surveillance, cancer prevention, and treatment.
Cancer genetics and the mechanisms behind clinical features are complex and require expertise for interpretation. We have gathered here ten general and comprehensible facts about Lynch syndrome the review article also highlights.
Lynch syndrome is the most prevalent hereditary cancer syndrome, with an estimated prevalence of 1 in 279, which would globally translate to 30 million people
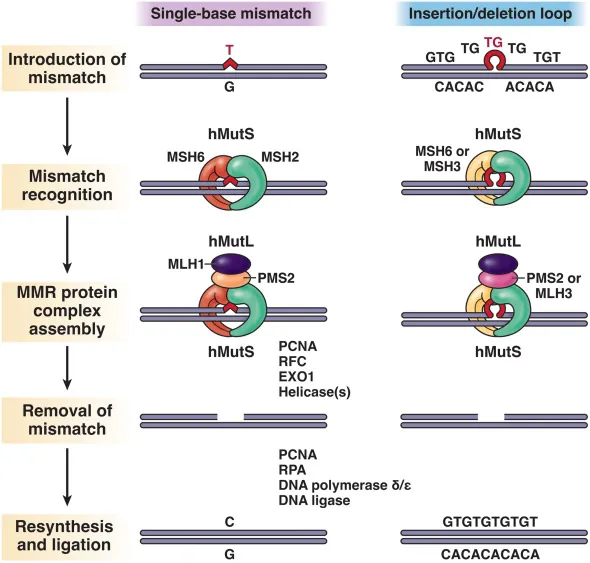
LS predisposes the affected persons to cancers, especially to colorectal cancer (CRC) and endometrial cancer (EC), because of an inherited defect in the DNA mismatch repair (MMR) mechanism.
Lynch syndrome is dominantly inherited
Individuals inherit their predisposing genetic variant from one parent and pass it on to their children with a 50% probability.
Researchers have discovered five genes associated with Lynch syndrome
Heterozygous germline variants in MMR genes MLH1, MSH2, MSH6, and PMS2, and the deletion of the 3´end of EPCAM (rare) are the identified underlying genetic causes for developing Lynch syndrome. PMS2 and MSH6 have demonstrated the highest population prevalence, whereas MLH1 and MSH2 are linked to the most increased susceptibility to developing cancer.
Lynch Syndrome associated cancer develops because of malfunction in the DNA mismatch repair (MMR)
Typically, the MMR mechanism corrects errors that arise during DNA replication and recombination, and maintains genomic stability in a cell. A functional MMR produces repair proteins, transports them to the nucleus, and forms protein complexes that perform the actual repair. In Lynch syndrome, an inherited gene alteration disrupts this function and predisposes to cancer.
Microsatellite instability MSI as a hallmark of Lynch syndrome cancers
Defective MMR (dMMR) results in length variation of short tandem nucleotide repeats, microsatellites, which is a hallmark of LS cancers and up to 30% of sporadic cancers.
The median age of cancer onset in LS is 45 years, about 20 years earlier than in sporadic cancers, although cancer risk may vary between the associated genes and sexes
Pathogenic variants in MLH1 and MSH2 produce a lifetime cancer risk of about 80% to both sexes. In contrast, the lifetime cancer risk in MSH6 carriers is 29% in males and 55% in females due to the increased risk for endometrial cancer.
The guidelines for cancer surveillance recommend early monitoring for Lynch syndrome individuals
The recommended age to start colorectal surveillance is 20-35, and 25-35 for gynecologic surveillance. Through increased understanding of the gene-associated differences in risk, clinical management guidelines are turning more gene-specific.
Lifestyle matters; physical activity and weight management help in cancer prevention
Increased physical activity and reduced body adiposity are associated with decreased cancer risk in the general population, and the same applies to LS.
Multiple guidelines recommend testing all new colorectal and endometrial cancers for selecting potential Lynch syndrome cases
Universal tumor screening is cost-efficient for selecting potential LS cases, and for overall cancer management after the checkpoint inhibition therapy became available for MMR deficient tumors. Immunohistochemical staining and microsatellite instability analysis are commonly used methods to detect deficient MMR protein expression and/or MSI in a tumor, respectively. Furthermore, MSI phenomenon offers a promising future approach for vaccine-based immunotherapy and immunoprevention of LS-associated cancers.
Efficient and early detection of Lynch syndrome becomes increasingly important, along with new modalities for cancer prevention
The major LS-cancers are associated with excellent survival, which is likely to reflect early detection and regular surveillance. However, the current tumor based approaches need to be supplemented with other approaches, such as functional methods capable of diagnosing heritable predisposition also in cases with clinical suspicion of LS but with no identifiable sequence changes, and in unaffected family members. LS CancerDiag provides a novel method, DiagMMR, to study MMR function in non-tumorous cells and recognizing Lynch syndrome without the necessity to find or interpret DNA variants.
To read the full article, please visit the Gastroenterology journal website
DNA Mismatch Repair Mechanism